The countdown has begun!
Do you have a medical device currently regulated under IVDD or a new medical device due for launch onto the EU IVD market?
If so, you could be living on the regulatory edge without realising it....
If you do not have your medical device regulated and authorised under IVDR by the relevant deadline, your medical device would be deemed to be non-compliant with EU law and potentially a threat to patient care and safety. As such, it would be immediately and permanently withdrawn from the EU market.
When did this all happen? What is the expectation for legacy and new medical devices?
On 26th May 2022 a huge, but expected, announcement broke into the scientific and medical device sector. A major shift and shakeup in the way IVDs (current and future) were going to be regulated on the EU market, from self-regulation through IVDD to mandatory evidence based regulation and accreditation through IVDR, following after a 5-year transitional period, was now what lay ahead of skeptical companies.
IVDs are now expected to transition from the self-regulating governance of the historic IVDD directive, that had been in place from 1998 (Diagnostic Directive of 1998 (98/79/EC or ‘IVDD’), if companies wish to continue to sell into, or become part of, of the EU IVD market. All current and emerging medical devices must now comply with the new and unknown lands of the In Vitro Diagnostic Medical Devices Regulation published in 2017 (Regulation 2017/746 or ’IVDR’).
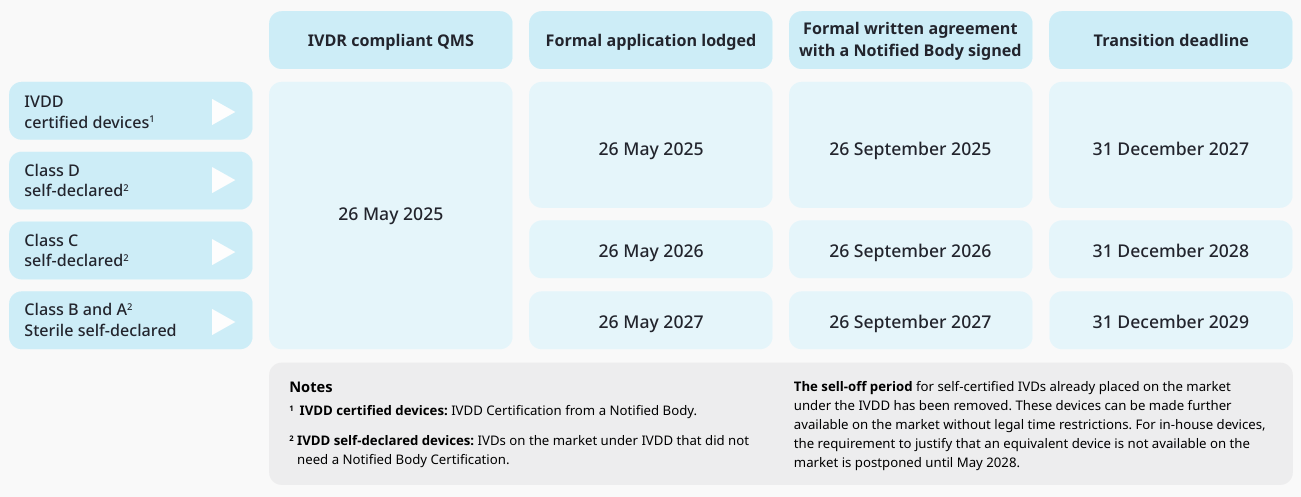
The IVDR deadlines have now been extended, have they not?
The next twist in the tale, comes with the extension of the original IVDR transition deadlines for only those medical devices that apply to additional specific conditions set out in the amended and published EU Regulation 2024/1860 on 9 July 2024. These changes were made to mitigate the potential risks of shortages of IVD medical devices caused by delays anticipated due to the complexity and sheer volume if devices expected transition to IVDR; whilst ensuring the continuation of patient care, without any compromise of safety.
What is the current status of IVD medical device regulations? Surely a lot have already gone through the IVDR regulation process?
Fast forward to the present day and it seems that not all companies are fully prepared for this essential update and are instead ‘living life on the edge’ with regards to the firm deadline outlined by Notified Bodies.
Of the estimated 50,000 IVD medical devices currently on the EU IVD market, only a small number are understood to have transitioned successfully to be certified under the IVDR certification process.
How can we best use the time until the Deadlines arrive?
PREPARE, PREPARE, PREPARE
Companies must effectively plan and prepare, to save unnecessary delays to IVDR submissions.
- Identify a Notified Body that covers your device.
- Get a writing team together of qualified individuals and/or external consultancy experts on-board early to provide regulatory advice and support. External consultants, such as Guy Howland Ltd, provide a cost-effective and efficient approach to the process, particularly due to time and internal resource constraints within companies. Templates can also be provided to ensure consistency across documentation and within the formal submission to the notified body.
- Prepare a realistic timeline for the IVDR submission. Assessments of documentation can only be made once all documentation has been submitted to the Notified Body. Allow time for reviews and questions. Notified Body reviews could have significant delays and therefore a significant impact on IVDR submission timelines if not anticipated and booked well in advance.
- Documentation must be well structured, complete, organized and fully accessible to facilitate rapid Notified Body review, with continual document updates.
- Submit your applications as early as possible to avoid the bottleneck.
- Keep updated with all current and relevant Notified Body guidance and documentation on IVDR submissions.
So, what do you need to do to avoid the cliff edge?
Act now, do not delay, understand the relevant legal deadlines and plan effectively to make the most of the time remaining to avoid any unexpected delays to authorisation/certification.
Ask the friendly, dedicated and knowledgeable consultancy experts at Guy Howland Ltd to help you, early in your planning and documentation journey, to ensure a successful IVDR submission to your notified body and the subsequent regulatory compliance.
We know the updated ins and outs for IVDR submissions and have handy templates to make the process as smooth as possible.
Contact us today to learn more!
References
Lubbers BR, Schilhabel A, Cobbaert CM, Gonzalez D, Dombrink I, Brüggemann M, Bitter WM, van Dongen JJM. The New EU Regulation on In Vitro Diagnostic Medical Devices: Implications and Preparatory Actions for Diagnostic Laboratories. Hemasphere. 2021 Apr 21;5(5):e568. doi: 10.1097/HS9.0000000000000568. PMID: 33898932; PMCID: PMC8061679.
BSI (Notified Body) Website: IVDR Transition Timelines Extended | BSI
Copyright © 2024 Guy Howland Ltd.
Web design by AB Business Solutions
We need your consent to load the translations
We use a third-party service to translate the website content that may collect data about your activity. Please review the details in the privacy policy and accept the service to view the translations.